Publications
Peer-reviewed publications published in the context of this project
-
Wednesday, Feb 3, 2021
Molecular Biology of the Cell
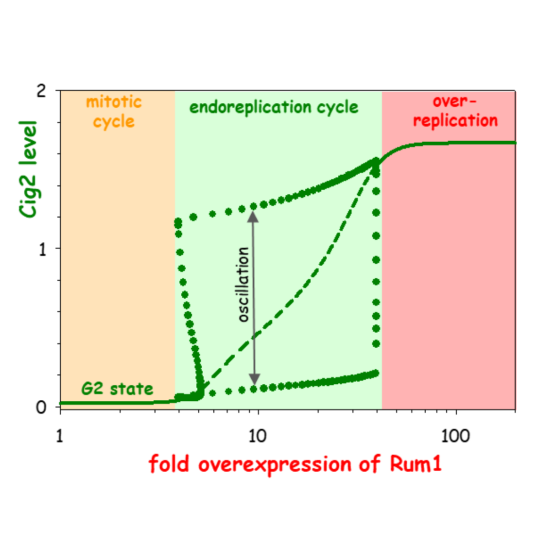
Computational Modelling of Chromosome Re-Replication in Mutant Strains of Fission Yeast
Bela Novak, John J. Tyson
-
AbstractTypically cells replicate their genome only once per division cycle, but under some circumstances, both natural and unnatural, cells synthesize an overabundance of DNA, either in a disorganized fashion (‘over-replication’) or by a systematic doubling of chromosome number (‘endoreplication’). These variations on the theme of DNA replication and division have been studied in strains of fission yeast, Schizosaccharomyces pombe, carrying mutations that interfere with the function of mitotic cyclin-dependent kinase (Cdk1:Cdc13) without impeding the roles of DNA-replication loading-factor (Cdc18) and S-phase cyclin-dependent kinase (Cdk1:Cig2). Some of these mutations support endoreplication, and some over-replication. In this paper, we propose a dynamical model of the interactions among the proteins governing DNA replication and cell division in fission yeast. By computational simulations of the mathematical model, we account for the observed phenotypes of these re-replicating mutants, and by theoretical analysis of the dynamical system, we provide insight into the molecular distinctions between over-replicating and endoreplicating cells. In case of induced over- production of regulatory proteins, our model predicts that cells first switch from normal mitotic cell cycles to growth-controlled endoreplication, and ultimately to disorganized over-replication, parallel to the slow increase of protein to very high levels.
-
Monday, Jan 4, 2021
Current Opinion in Cell Biology
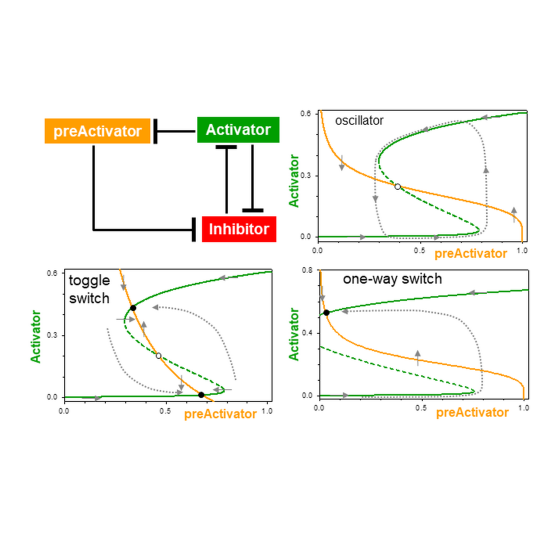
Mechanisms of Signalling-Memory Governing Progression Through the Eukaryotic Cell Cycle
Bela Novak, John J. Tyson
-
AbstractAs cells pass through each replication-division cycle, they must be able to postpone further progression if they detect any threats to genome integrity, such as DNA damage or misaligned chromosomes. Once a 'decision' is made to proceed, the cell unequivocally enters into a qualitatively different biochemical state, which makes the transitions from one cell cycle phase to the next switch-like and irreversible. Each transition is governed by a unique signalling network; nonetheless, they share a common characteristic of bistable behaviour, a hallmark of molecular memory devices. Comparing the cell cycle signalling mechanisms acting at the restriction point, G1/S, G2/M and meta-to-anaphase transitions, we deduce a generic network motif of coupled positive and negative feedback loops underlying each transition.
-
Thursday, Apr 30, 2020
Trends in Cell Biology
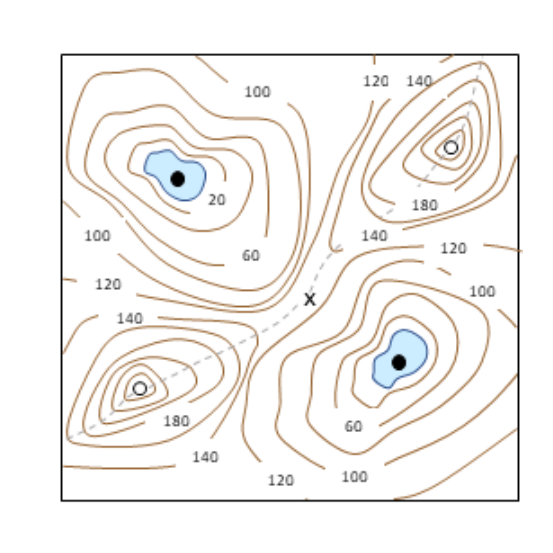
A Dynamical Paradigm for Molecular Cell Biology
Bela Novak, John J. Tyson
-
AbstractThe driving passion of molecular cell biologists is to understand the molecular mechanisms that control important aspects of cell physiology, but this ambition is often limited by the wealth of molecular details currently known about these mechanisms. Their complexity overwhelms our intuitive notions of how molecular regulatory networks might respond under normal and stressful conditions. To make progress we need a new paradigm for connecting molecular biology to cell physiology. We suggest an approach that uses precise mathematical methods to associate the qualitative features of dynamical systems, as conveyed by ‘bifurcation diagrams’, with ‘signal- response’ curves measured by cell biologists.
-
Thursday, Apr 30, 2020
EMBO Journal
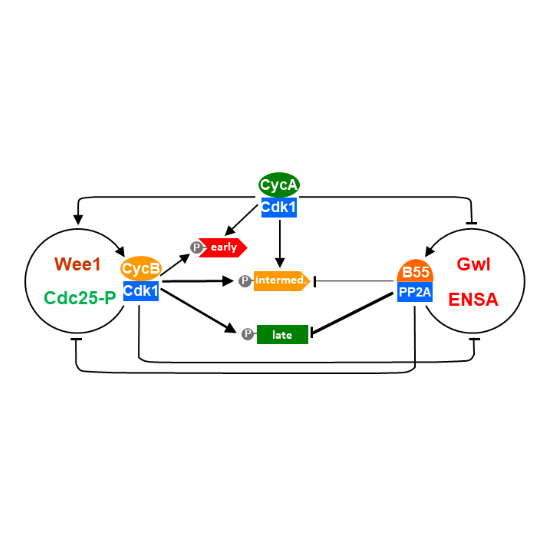
Cyclin A Triggers Mitosis Either via the Greatwall Kinase Pathway or Cyclin B
Nadia Hegarat, Adrijana Crncec, Maria F Suarez Peredo Rodriguez, Fabio Echegaray Iturra, Yan Gu, Oliver Busby, Paul F Lang, Alexis R Barr, Chris Bakal, Masato T Kanemaki, Angus I Lamond, Bela Novak, Tony Ly, Helfrid Hochegger
-
AbstractTwo mitotic cyclin types, cyclin A and B, exist in higher eukaryotes, but their specialised functions in mitosis are incompletely understood. Using degron tags for rapid inducible protein removal, we analyse how acute depletion of these proteins affects mitosis. Loss of cyclin A in G2-phase prevents mitotic entry. Cells lacking cyclin B can enter mitosis and phosphorylate most mitotic proteins, because of parallel PP2A:B55 phosphatase inactivation by Greatwall kinase. The final barrier to mitotic establishment corresponds to nuclear envelope breakdown, which requires a decisive shift in the balance of cyclin-dependent kinase Cdk1 and PP2A:B55 activity. Beyond this point, cyclin B/Cdk1 is essential for phosphorylation of a distinct subset of mitotic Cdk1 substrates that are essential to complete cell division. Our results identify how cyclin A, cyclin B and Greatwall kinase coordinate mitotic progression by increasing levels of Cdk1-dependent substrate phosphorylation.
-
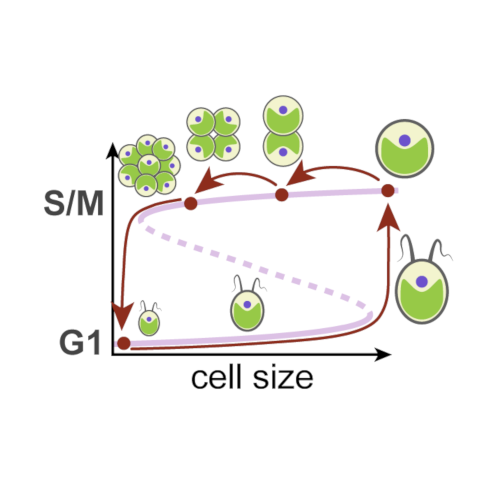
Thursday, Jan 9, 2020
Current Biology
A single light-responsive sizer can control multiple-fission cycles in Chlamydomonas
Stefan Heldt, John J. Tyson, Frederick R. Cross, Bela Novak
-
AbstractMost eukaryotic cells execute binary division after each mass doubling in order to maintain size homeostasis by coordinating cell growth and division. By contrast, the photosynthetic green alga Chlamydomonas can grow more than 8-fold during daytime and then, at night, undergo rapid cycles of DNA replication, mitosis, and cell division, producing up to 16 daughter cells. Here, we propose a mechanistic model for multiple-fission cycles and cell-size control in Chlamydomonas. The model comprises a light-sensitive and size-dependent biochemical toggle switch that acts as a sizer, guarding transitions into and exit from a phase of cell-division cycle oscillations. This simple “sizer-oscillator” arrangement reproduces the experimentally observed features of multiple-fission cycles and the response of Chlamydomonas cells to different light-dark regimes. Our model also makes specific predictions about the size dependence of the time of onset of cell division after cells are transferred from light to dark conditions, and we confirm these predictions by single-cell experiments. Collectively, our results provide a new perspective on the concept of a “commitment point” during the growth of Chlamydomonas cells and hint at intriguing similarities of cell-size control in different eukaryotic lineages.
-
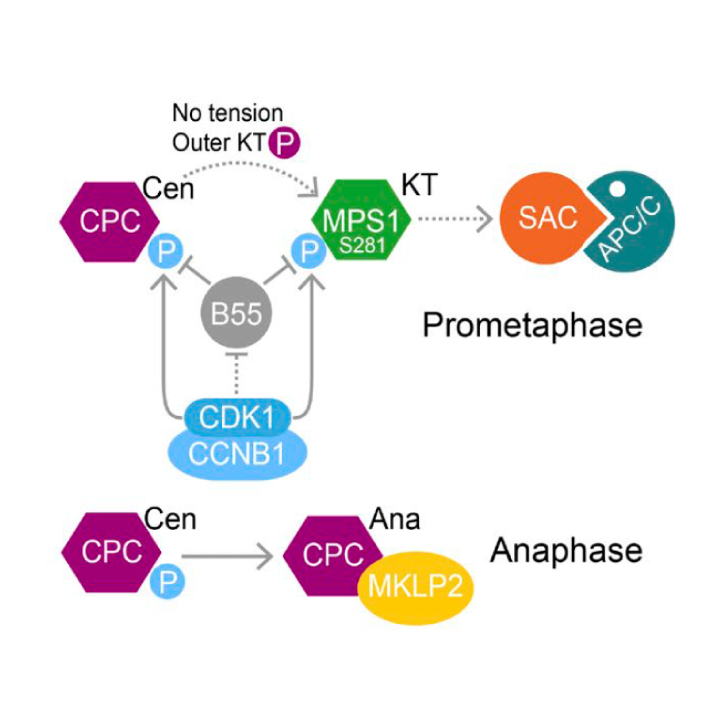
Wednesday, Jan 23, 2019
Journal of Cell Biology
CDK1-CCNB1 creates a spindle checkpoint–permissive state by enabling MPS1 kinetochore localization
Daniel Hayward, Tatiana Alfonso-Pérez, Michael J. Cundell, Michael Hopkins, James Holder, James Bancroft, Lukas H. Hutter, Bela Novak, Francis A. Barr, Ulrike Gruneberg
-
AbstractSpindle checkpoint signaling is initiated by recruitment of the kinase MPS1 to unattached kinetochores during mitosis. We show that CDK1-CCNB1 and a counteracting phosphatase PP2A-B55 regulate the engagement of human MPS1 with unattached kinetochores by controlling the phosphorylation status of S281 in the kinetochore-binding domain. This regulation is essential for checkpoint signaling, since MPS1S281A is not recruited to unattached kinetochores and fails to support the recruitment of other checkpoint proteins. Directly tethering MPS1S281A to the kinetochore protein Mis12 bypasses this regulation and hence the requirement for S281 phosphorylation in checkpoint signaling. At the metaphase–anaphase transition, MPS1 S281 dephosphorylation is delayed because PP2A-B55 is negatively regulated by CDK1-CCNB1 and only becomes fully active once CCNB1 concentration falls below a characteristic threshold. This mechanism prolongs the checkpoint-responsive period when MPS1 can localize to kinetochores and enables a response to late-stage spindle defects. By acting together, CDK1-CCNB1 and PP2A-B55 thus create a spindle checkpoint–permissive state and ensure the fidelity of mitosis.
-
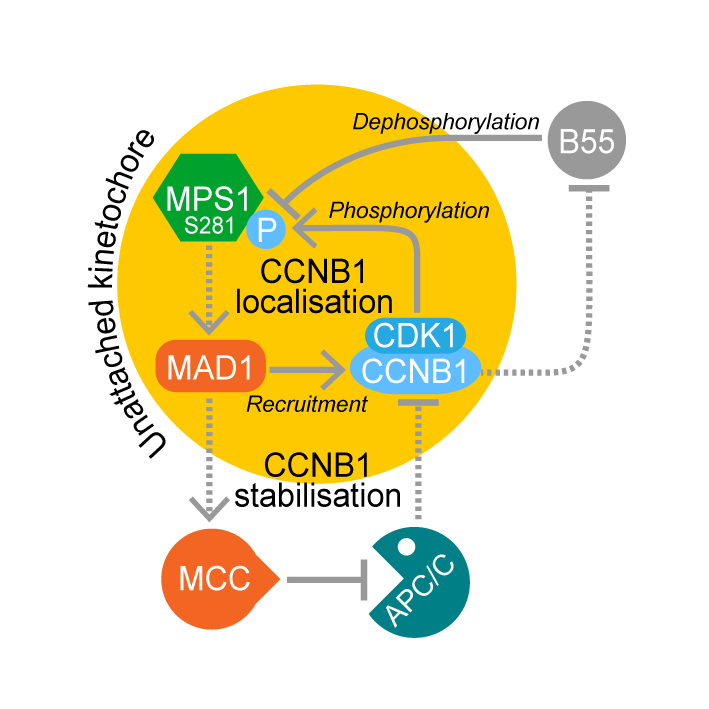
Wednesday, Jan 23, 2019
Journal of Cell Biology
MAD1-dependent recruitment of CDK1-CCNB1 to kinetochores promotes spindle checkpoint signaling
Tatiana Alfonso-Pérez, Daniel Hayward, James Holder, Ulrike Gruneberg, Francis A. Barr
-
AbstractCyclin B–dependent kinase (CDK1-CCNB1) promotes entry into mitosis. Additionally, it inhibits mitotic exit by activating the spindle checkpoint. This latter role is mediated through phosphorylation of the checkpoint kinase MPS1 and other spindle checkpoint proteins. We find that CDK1-CCNB1 localizes to unattached kinetochores and like MPS1 is lost from these structures upon microtubule attachment. This suggests that CDK1-CCNB1 is an integral component and not only an upstream regulator of the spindle checkpoint pathway. Complementary proteomic and cell biological analysis demonstrate that the spindle checkpoint protein MAD1 is one of the major components of CCNB1 complexes, and that CCNB1 is recruited to unattached kinetochores in an MPS1-dependent fashion through interaction with the first 100 amino acids of MAD1. This MPS1 and MAD1-dependent pool of CDK1-CCNB1 creates a positive feedback loop necessary for timely recruitment of MPS1 to kinetochores during mitotic entry and for sustained spindle checkpoint arrest. CDK1-CCNB1 is therefore an integral component of the spindle checkpoint, ensuring the fidelity of mitosis.
-
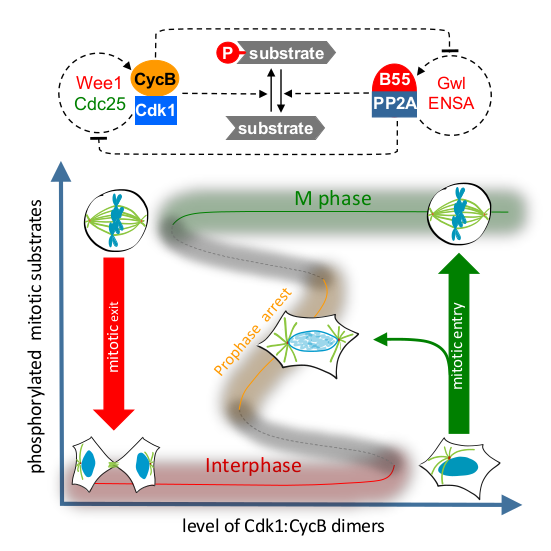
Thursday, Nov 15, 2018
Current Biology
Two Interlinked Bistable Switches Govern Mitotic Control in Mammalian Cells
Scott Rata, Maria F. Suarez Peredo Rodriguez, Stephy Joseph, Nisha Peter, Fabio E. Iturra, Fengwei Yang, Anotida Madzvamuse, Jan G. Ruppert, Kumiko Samejima, Melpomeni Platani, Monica Alvarez-Fernandez, Marcos Malumbres, William C. Earnshaw, Bela Novak, Helfrid Hochegger
-
AbstractDistinct protein phosphorylation levels in interphase and M phase require tight regulation of Cdk1 activity [1,2]. A bistable switch, based on positive feedback in the Cdk1 activation loop, has been proposed to generate different thresholds for transitions between these cell-cycle states [3–5]. Recently, the activity of the major Cdk1-counteracting phosphatase, PP2A:B55, has also been found to be bistable due to Greatwall kinase-dependent regulation [6]. However, the interplay of the regulation of Cdk1 and PP2A:B55 in vivo remains unexplored. Here, we combine quantitative cell biology assays with mathematical modeling to explore the interplay of mitotic kinase activation a nd phosphatase inactivation in human cells. By measuring mitotic entry and exit thresholds using ATP-analog-sensitive Cdk1 mutants, we find evidence that the mitotic switch displays hysteresis and bistability, responding differentially to Cdk1 inhibition in the mitotic and interphase states. Cdk1 activation by Wee1/Cdc25 feedback loops and PP2A:B55 inactivation by Greatwall independently contributes to this hysteretic switch system. However, elimination of both Cdk1 and PP2A:B55 inactivation fully abrogates bistability, suggesting that hysteresis is an emergent property of mutual inhibition between the Cdk1 and PP2A:B55 feedback loops. Our model of the two interlinked feedback systems predicts an intermediate but hidden steady state between interphase and M phase. This could be verified experimentally by Cdk1 inhibition during mitotic entry, supporting the predictive value of our model. Furthermore, we demonstrate that dual inhibition of Wee1 and Gwl kinases causes loss of cell-cycle memory and synthetic lethality, which could be further exploited therapeutically.
-
Wednesday, Oct 24, 2018
PLOS Computational Biology
Dilution and titration of cell-cycle regulators may control cell size in budding yeast
Stefan Heldt, Reece Lunstone, John J. Tyson, Bela Novak
-
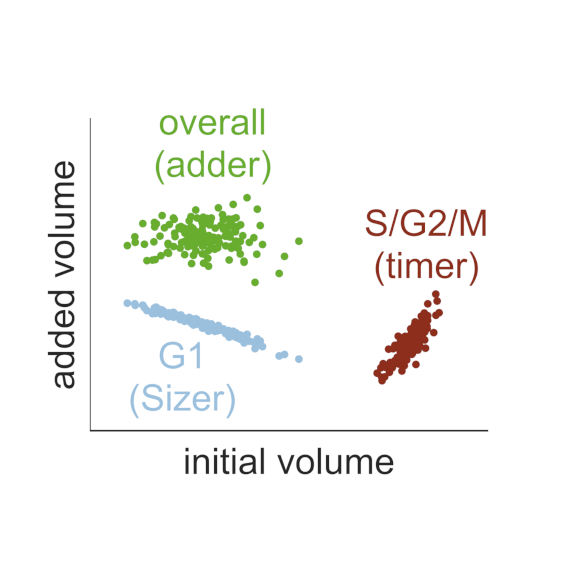
AbstractThe size of a cell sets the scale for all biochemical processes within it, thereby affecting cellular fitness and survival. Hence, cell size needs to be kept within certain limits and relatively constant over multiple generations. However, how cells measure their size and use this information to regulate growth and division remains controversial. Here, we present two mechanistic mathematical models of the budding yeast (S. cerevisiae) cell cycle to investigate competing hypotheses on size control: inhibitor dilution and titration of nuclear sites. Our results suggest that an inhibitor-dilution mechanism, in which cell growth dilutes the transcriptional inhibitor Whi5 against the constant activator Cln3, can facilitate size homeostasis. This is achieved by utilising a positive feedback loop to establish a fixed size threshold for the START transition, which efficiently couples cell growth to cell cycle progression. Yet, we show that inhibitor dilution cannot reproduce the size of mutants that alter the cell’s overall ploidy and WHI5 gene copy number. By contrast, size control through titration of Cln3 against a constant number of genomic binding sites for the transcription factor SBF recapitulates both size homeostasis and the size of these mutant strains. Moreover, this model produces an imperfect ‘sizer’ behaviour in G1 and a ‘timer’ in S/G2/M, which combine to yield an ‘adder’ over the whole cell cycle; an observation recently made in experiments. Hence, our model connects these phenomenological data with the molecular details of the cell cycle, providing a systems-level perspective of budding yeast size control.
-
Wednesday, Aug 1, 2018
Molecular Cell
DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation
Bennie Lemmens, Nadia Hegarat, Karen Akopyan, Joan Sala-Gaston, Jiri Bartek, Helfrid Hochegger, Arne Lindqvist
-
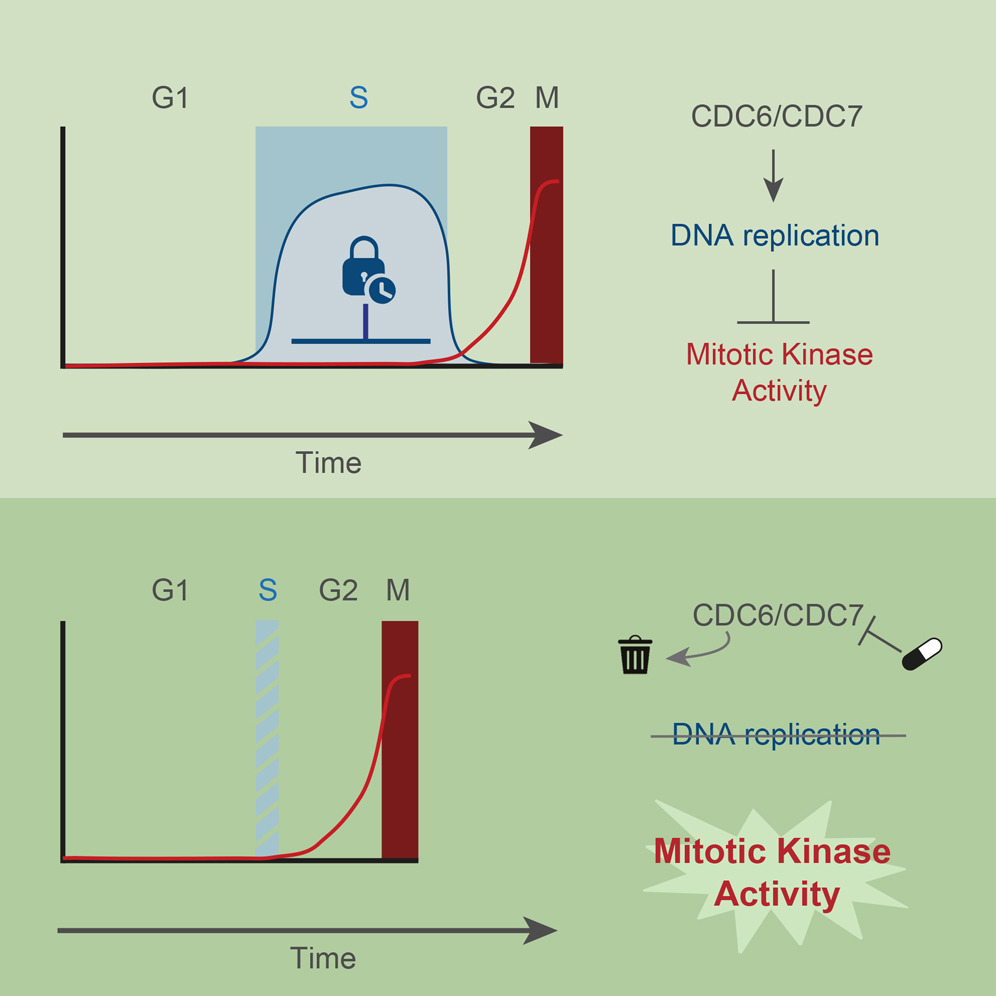
AbstractTo maintain genome stability, cells need to replicate their DNA before dividing. Upon completion of bulk DNA synthesis, the mitotic kinases CDK1 and PLK1 become active and drive entry into mitosis. Here, we have tested the hypothesis that DNA replication determines the timing of mitotic kinase activation. Using an optimized double-degron system, together with kinase inhibitors to enforce tight inhibition of key proteins, we find that human cells unable to initiate DNA replication prematurely enter mitosis. Preventing DNA replication licensing and/or firing causes prompt activation of CDK1 and PLK1 in S phase. In the presence of DNA replication, inhibition of CHK1 and p38 leads to premature activation of mitotic kinases, which induces severe replication stress. Our results demonstrate that, rather than merely a cell cycle output, DNA replication is an integral signaling component that restricts activation of mitotic kinases. DNA replication thus functions as a brake that determines cell cycle duration.
-
Thursday, Feb 22, 2018
Current Opinion in Systems Biology
Genome Stability during Cell Proliferation: A Systems Analysis of the Molecular Mechanisms Controlling Progression through the Eukaryotic Cell Cycle
Bela Novak, Stefan Heldt, John J. Tyson
-
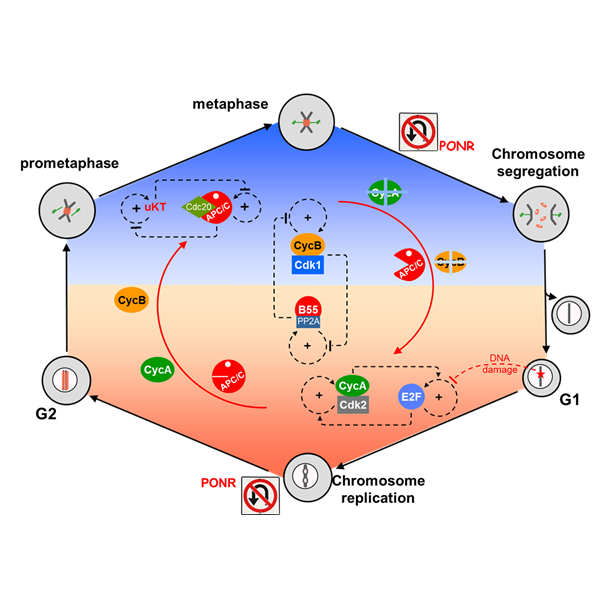
AbstractWell-nourished cells in a favorable environment (well supplied with growth factors, cytokines, and/or hormones and free from stresses, ionizing radiation, etc.) will grow, replicate their genome, and divide into two daughter cells, fully prepared to repeat the process. This cycle of DNA replication and division underlies all aspects of biological growth, reproduction, repair and development. As such, it is essential that the cell’s genome be guarded against damage during the replication/division process, lest the error(s) be irrevocably passed down to all future generations of progeny. Hence, cell cycle progression is closely guarded against major sources of errors, in particular DNA damage and misalignment of replicated chromosomes on the mitotic spindle. In this review article we examine closely the molecular mechanisms that maintain genomic integrity during the cell division cycle, and we find an unexpected and intriguing arrangement of concatenated and nested bistable toggle switches. The topology of the network seems to play crucial roles in maintaining the stability of the genome during cell proliferation.
-
Wednesday, Feb 21, 2018
Proceedings of the National Academy of Sciences
A comprehensive model for the proliferation-quiescence decision in response to endogenous DNA damage in human cells
Stefan Heldt, Alexis Barr, Sam Cooper, Chris Bakal, Bela Novak
-
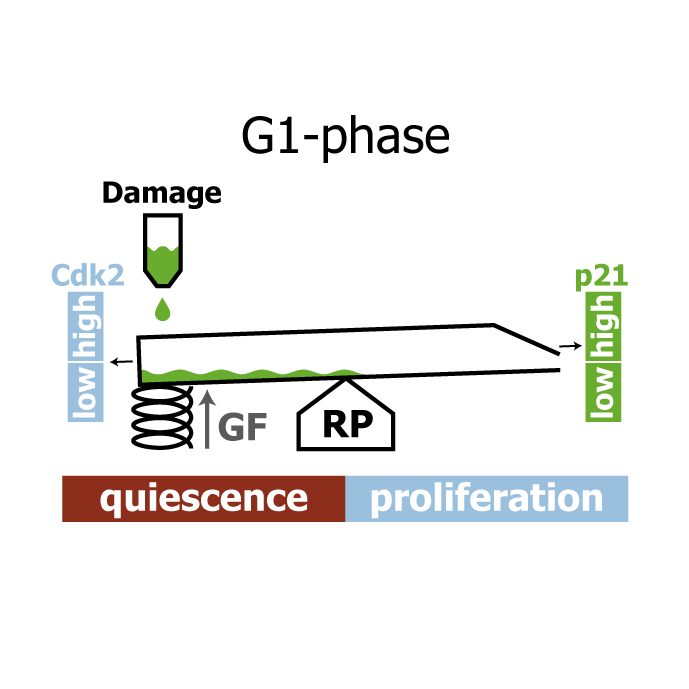
AbstractHuman cells that suffer mild DNA damage can enter a reversible state of growth arrest known as quiescence. This decision to temporarily exit the cell cycle is essential to prevent the propagation of mutations and most cancer cells harbour defects in the underlying control system. Here we present a mechanistic mathematical model to study the proliferation-quiescence decision in non-transformed human cells. We show that two bistable switches, the restriction point (RP) and the G1/S transition, mediate this decision by integrating DNA damage and mitogen signals. In particular, our data suggests that the cyclin-dependent kinase inhibitor p21 (Cip1/Waf1), which is expressed in response to DNA damage, promotes quiescence by blocking positive feedback loops that facilitate G1 progression downstream of serum stimulation. Intriguingly, cells exploit bistability in the RP to convert graded p21 and mitogen signals into an all-or-nothing cell cycle response. The same mechanism creates a window of opportunity where G1 cells that have passed the RP can revert to quiescence if exposed to DNA damage. We present experimental evidence that cells gradually lose this ability to revert to quiescence as they progress through G1 and that the onset of rapid p21 degradation at the G1/S transition prevents this response altogether, insulating S-phase from mild, endogenous DNA damage. Thus, two bistable switches conspire in the early cell cycle to provide both sensitivity and robustness to external stimuli.
-
Thursday, Nov 9, 2017
Molecular Biology of the Cell
Cell cycle transitions: a common role for stoichiometric inhibitors
Michael Hopkins, John Tyson, Bela Novak
-
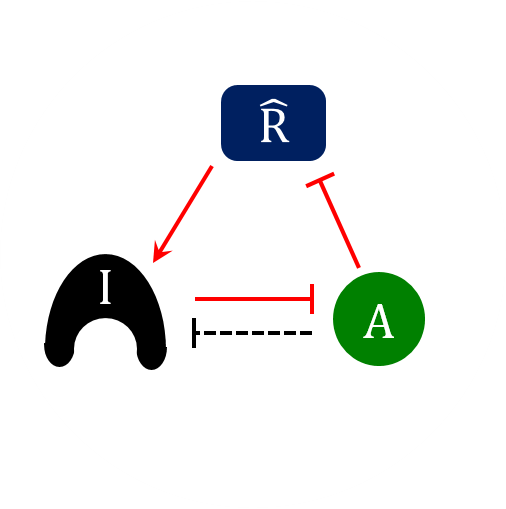
AbstractThe cell division cycle is the process by which eukaryotic cells replicate their chromosomes and partition them to two daughter cells. To maintain the integrity of the genome, proliferating cells must be able to block progression through the division cycle at key transition points (called “checkpoints”) if there have been problems in the replication of the chromosomes or their biorientation on the mitotic spindle. These checkpoints are governed by protein-interaction networks, composed of phase-specific cell-cycle activators and inhibitors. Examples include Cdk1:Clb5 and its inhibitor Sic1 at the G1/S checkpoint in budding yeast, APC:Cdc20 and its inhibitor MCC at the mitotic checkpoint, and PP2A:B55 and its inhibitor, alpha-endosulfine, at the mitotic-exit checkpoint. Each of these inhibitors is a substrate as well as a stoichiometric inhibitor of the cell-cycle activator. Because the production of each inhibitor is promoted by a regulatory protein that is itself inhibited by the cell-cycle activator, their interaction network presents a regulatory motif characteristic of a “feedback-amplified domineering substrate” (FADS). We describe how the FADS motif responds to signals in the manner of a bistable toggle switch, and then we discuss how this toggle switch accounts for the abrupt and irreversible nature of three specific cell-cycle checkpoints.
-
Tuesday, Sep 19, 2017
Cell Cycle
Interlinked bistable mechanisms generate robust mitotic transitions
Lukas Hutter, Scott Rata, Helfrid Hochegger, Bela Novak
-
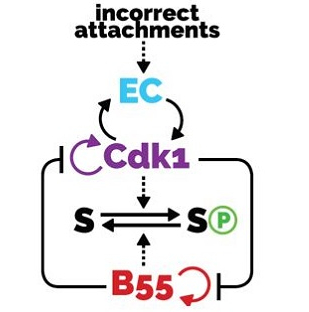
AbstractThe transitions between phases of the cell cycle have evolved to be robust and switch-like, which ensures temporal separation of DNA replication, sister chromatid separation, and cell division. Mathematical models describing the biochemical interaction networks of cell cycle regulators attribute these properties to underlying bistable switches, which inherently generate robust, switch-like, and irreversible transitions between states. We have recently presented new mathematical models for two control systems that regulate crucial transitions in the cell cycle: mitotic entry and exit and the mitotic checkpoint.Each of the two control systems is characterized by two interlinked bistable switches. In the case of mitotic checkpoint control, these switches are mutually activating, whereas in the case of the mitotic entry/exit network, the switches are mutually inhibiting. In this Perspective we describe the qualitative features of these regulatory motifs and show that having two interlinked bistable mechanisms further enhances robustness and irreversibility. We speculate that these network motifs also underlie other cell cycle transitions and cellular transitions between distinct biochemical states.
-

Tuesday, Jun 20, 2017
Bioinformatics
NucliTrack: An integrated nuclei tracking application
Sam Cooper, Alexis Barr, Robert Glen, Chris Bakal
-
AbstractLive imaging studies give unparalleled insight into dynamic single cell behaviours and fate decisions. However, the challenge of reliably tracking single cells over long periods of time limits both the throughput and ease with which such studies can be performed. Here, we present NucliTrack, a cross platform solution for automatically segmenting, tracking and extracting features from fluorescently-labelled nuclei. NucliTrack performs similarly to other state-of-the-art cell tracking algorithms, but NucliTrack’s interactive, graphical interface makes it significantly more user friendly.
-
Monday, May 22, 2017
Current Biology
APC/C:Cdh1 Enables Removal of Shugoshin-2 from the Arms of Bivalent Chromosomes by Moderating Cyclin-Dependent Kinase Activity
Ahmed Rattani, Randy Ballesteros Mejia, Katherine Roberts, Maurici B. Roig, Jonathan Godwin, Michael Hopkins, Manuel Eguren, Luis Sanchez-Pulido, Elwy Okaz, Sugako Ogushi, Magda Wolna, Jean Metson, Alberto M. Pendas, Marcos Malumbres, Bela Novak, Mary Herbert, Kim Nasmyth
-
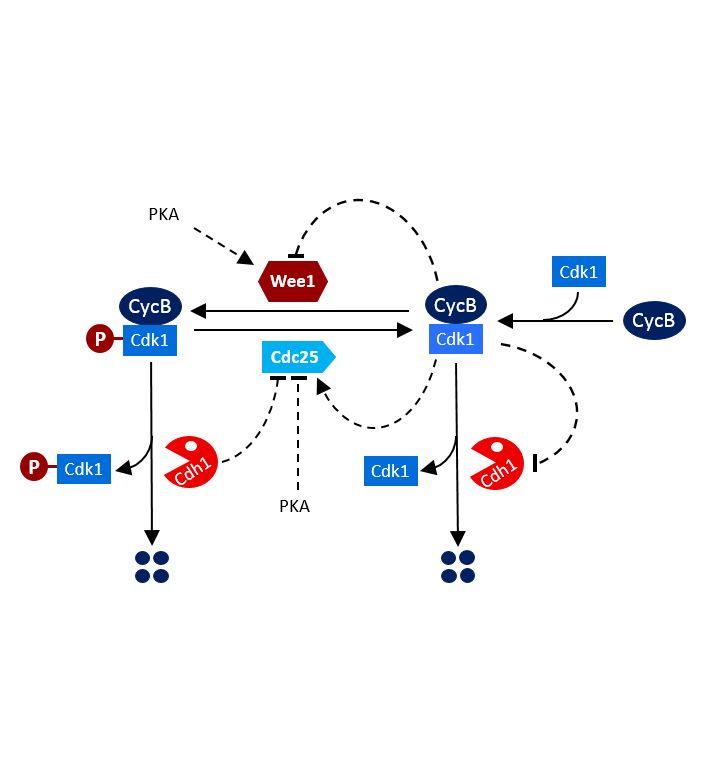
AbstractIn mammalian females, germ cells remain arrested as primordial follicles. Resumption of meiosis is heralded by germinal vesicle breakdown, condensation of chromosomes, and their eventual alignment on metaphase plates. At the first meiotic division, anaphase-promoting complex/cyclosome associated with Cdc20 (APC/CCdc20) activates separase and thereby destroys cohesion along chromosome arms. Because cohesion around centromeres is protected by shugoshin-2, sister chromatids remain attached through centromeric/pericentromeric cohesin. We show here that, by promoting proteolysis of cyclins and Cdc25B at the germinal vesicle (GV) stage, APC/C associated with the Cdh1 protein (APC/CCdh1) delays the increase in Cdk1 activity, leading to germinal vesicle breakdown (GVBD). More surprisingly, by moderating the rate at which Cdk1 is activated following GVBD, APC/CCdh1 creates conditions necessary for the removal of shugoshin-2 from chromosome arms by the Aurora B/C kinase, an event crucial for the efficient resolution of chiasmata.
-
Monday, Mar 20, 2017
Nature Communications
DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression
Alexis Barr, Samuel Cooper, Stefan Heldt, Francesca Butera, Henriette Stoy, Jörg Mansfeld, Bela Novak, Chris Bakal
-
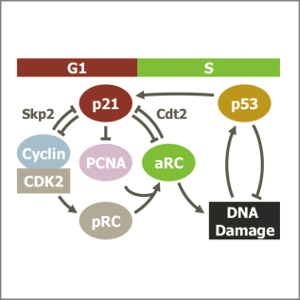
AbstractFollowing DNA damage caused by exogenous sources, such as ionising radiation, the tumour suppressor p53 mediates cell cycle arrest via expression of the CDK inhibitor, p21. However, the role of p21 in maintaining genomic stability in the absence of exogenous DNA damaging agents is unclear. Here, using live single cell measurements of p21 protein in proliferating cultures, we show that naturally-occurring DNA damage incurred over S-phase causes p53-dependent accumulation of p21 during mother G2- and daughter G1-phases. High p21 levels mediate G1 arrest via CDK inhibition, yet lower levels have no impact on G1 progression, and the ubiquitin ligases CRL4Cdt2 and SCFSkp2 couple to degrade p21 prior to the G1/S transition. Mathematical modelling reveals that a bistable switch, created by CRL4Cdt2, promotes irreversible S-phase entry by keeping p21 levels low, preventing premature S-phase exit upon DNA damage. Thus, we characterise how p21 regulates the proliferation-quiescence decision to maintain genomic stability.
-
Wednesday, Nov 23, 2016
Current Biology
Two Bistable Switches Govern M Phase Entry
Satoru Mochida, Scott Rata, Hirotsugu Hino, Takeharu Nagai, Bela Novak
-
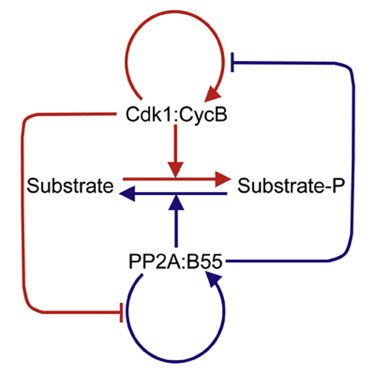
AbstractThe abrupt and irreversible transition from interphase to M phase is essential to separate DNA replication from chromosome segregation. This transition requires the switch-like phosphorylation of hundreds of proteins by the cyclin-dependent kinase 1 (Cdk1):cyclin B (CycB) complex. Previous studies have ascribed these switch-like phosphorylations to the auto-activation of Cdk1:CycB through the removal of inhibitory phosphorylations on Cdk1-Tyr15 [1 and 2]. The positive feedback in Cdk1 activation creates a bistable switch that makes mitotic commitment irreversible [2, 3 and 4]. Here, we surprisingly find that Cdk1 auto-activation is dispensable for irreversible, switch-like mitotic entry due to a second mechanism, whereby Cdk1:CycB inhibits its counteracting phosphatase (PP2A:B55). We show that the PP2A:B55-inhibiting Greatwall (Gwl)-endosulfine (ENSA) pathway is both necessary and sufficient for switch-like phosphorylations of mitotic substrates. Using purified components of the Gwl-ENSA pathway in a reconstituted system, we found a sharp Cdk1 threshold for phosphorylation of a luminescent mitotic substrate. The Cdk1 threshold to induce mitotic phosphorylation is distinctly higher than the Cdk1 threshold required to maintain these phosphorylations—evidence for bistability. A combination of mathematical modeling and biochemical reconstitution show that the bistable behavior of the Gwl-ENSA pathway emerges from its mutual antagonism with PP2A:B55. Our results demonstrate that two interlinked bistable mechanisms provide a robust solution for irreversible and switch-like mitotic entry.
-
Monday, Aug 22, 2016
Journal of Cell Biology
A PP2A-B55 recognition signal controls substrate dephosphorylation kinetics during mitotic exit
Michael Cundell, Lukas Hutter, Ricardo Nunes-Bastos, Elena Poser, James Holder, Shabaz Mohammed, Bela Novak, Francis Barr
-
AbstractPP2A-B55 is one of the major phosphatases regulating cell division. Despite its importance for temporal control during mitotic exit, how B55 substrates are recognized and differentially dephosphorylated is unclear. Using phosphoproteomics combined with kinetic modeling to extract B55-dependent rate constants, we have systematically identified B55 substrates and assigned their temporal order in mitotic exit. These substrates share a bipartite polybasic recognition determinant (BPR) flanking a Cdk1 phosphorylation site. Experiments and modeling show that dephosphorylation rate is encoded into B55 substrates, including its inhibitor ENSA, by cooperative action of basic residues within the BPR. A complementary acidic surface on B55 decodes this signal, supporting a cooperative electrostatic mechanism for substrate selection. A further level of specificity is encoded into B55 substrates because B55 displays selectivity for phosphothreonine. These simple biochemical properties, combined with feedback control of B55 activity by the phosphoserine-containing substrate/inhibitor ENSA, can help explain the temporal sequence of events during exit from mitosis.
-
Friday, May 27, 2016
BioEssays
Bistability of mitotic entry and exit switches during open mitosis in mammalian cells
Nadia Hegarat, Scott Rata, Helfrid Hochegger
-
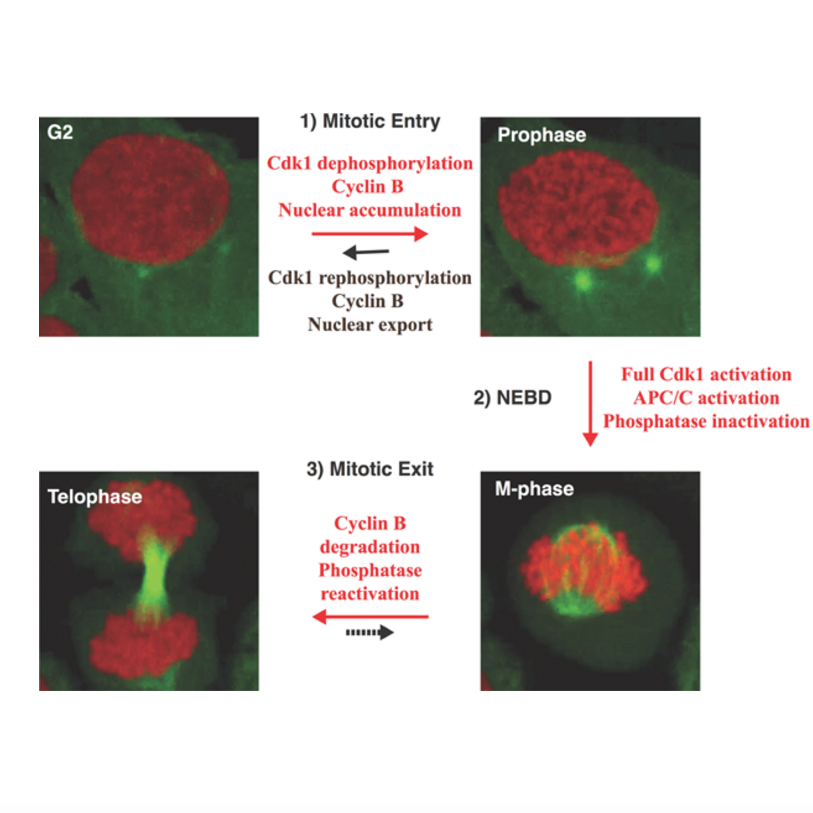
AbstractMitotic entry and exit are switch-like transitions that are driven by the activation and inactivation of Cdk1 and mitotic cyclins. This simple on/off reaction turns out to be a complex interplay of various reversible reactions, feedback loops, and thresholds that involve both the direct regulators of Cdk1 and its counteracting phosphatases. In this review, we summarize the interplay of the major components of the system and discuss how they work together to generate robustness, bistability, and irreversibility. We propose that it may be beneficial to regard the entry and exit reactions as two separate reversible switches that are distinguished by differences in the state of phosphatase activity, mitotic proteolysis, and a dramatic rearrangement of cellular components after nuclear envelope breakdown, and discuss how the major Cdk1 activity thresholds could be determined for these transitions.
-
Monday, Feb 8, 2016
Current Biology
Nutritional Control of Cell Size by the Greatwall-Endosulfine-PP2A·B55 Pathway
Nathalia Chica, Ana Rozalen, Livia Perez-Hidalgo, Angela Rubio, Bela Novak, Sergio Moreno
-
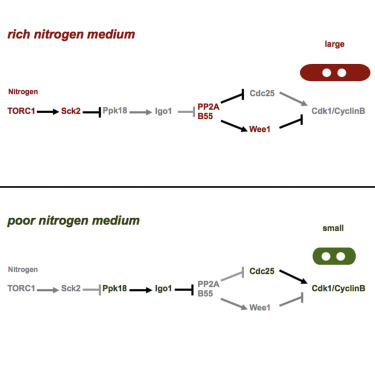
AbstractProliferating cells adjust their cell size depending on the nutritional environment. Cells are large in rich media and small in poor media. This physiological response has been demonstrated in both unicellular and multicellular organisms. Here we show that the greatwall-endosulfine (Ppk18-Igo1 in fission yeast) pathway couples the nutritional environment to the cell-cycle machinery by regulating the activity of PP2A·B55. In the presence of nutrients, greatwall (Ppk18) protein kinase is inhibited by TORC1 and PP2A·B55 is active. High levels of PP2A·B55 prevent the activation of mitotic Cdk1·Cyclin B, and cells increase in size in G2 before they undergo mitosis. When nutrients are limiting, TORC1 activity falls off, and the activation of greatwall (Ppk18) leads to the phosphorylation of endosulfine (Igo1) and inhibition of PP2A·B55, which in turn allows full activation of Cdk1·CyclinB and entry into mitosis with a smaller cell size. Given the conservation of this pathway, it is reasonable to assume that this mechanism operates in higher eukaryotes, as well.
-
Wednesday, Jan 27, 2016
Cell Systems
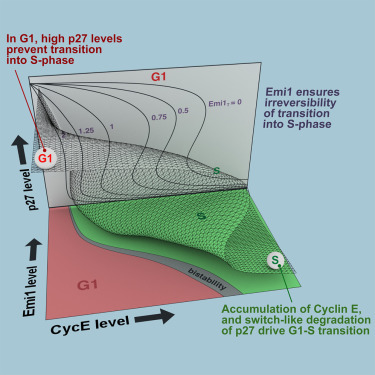
A Dynamical Framework for the All-or-None G1/S Transition
Alexis Barr, Stefan Heldt, Tongli Zhang, Chris Bakal, Bela Novak
-
AbstractThe transition from G1 into DNA replication (S phase) is an emergent behavior resulting from dynamic and complex interactions between cyclin-dependent kinases (Cdks), Cdk inhibitors (CKIs), and the anaphase-promoting complex/cyclosome (APC/C). Understanding the cellular decision to commit to S phase requires a quantitative description of these interactions. We apply quantitative imaging of single human cells to track the expression of G1/S regulators and use these data to parametrize a stochastic mathematical model of the G1/S transition. We show that a rapid, proteolytic, double-negative feedback loop between Cdk2:Cyclin and the Cdk inhibitor p27Kip1 drives a switch-like entry into S phase. Furthermore, our model predicts that increasing Emi1 levels throughout S phase are critical in maintaining irreversibility of the G1/S transition, which we validate using Emi1 knockdown and live imaging of G1/S reporters. This work provides insight into the general design principles of the signaling networks governing the temporally abrupt transitions between cell-cycle phases.
-
Thursday, Oct 8, 2015
Cell Reports
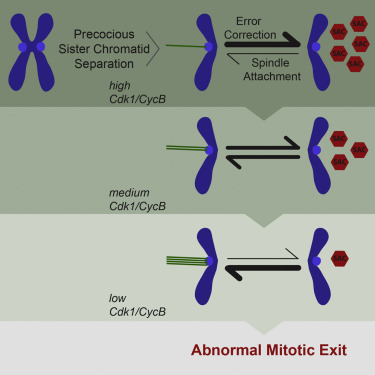
Premature Sister Chromatid Separation Is Poorly Detected by the Spindle Assembly Checkpoint as a Result of System-Level Feedback
Mihailo Mirkovic, Lukas Hutter, Bela Novak, Raquel Oliveira
-
AbstractSister chromatid cohesion, mediated by the cohesin complex, is essential for faithful mitosis. Nevertheless, evidence suggests that the surveillance mechanism that governs mitotic fidelity, the spindle assembly checkpoint (SAC), is not robust enough to halt cell division when cohesion loss occurs prematurely. The mechanism behind this poor response is not properly understood. Using developing Drosophila brains, we show that full sister chromatid separation elicits a weak checkpoint response resulting in abnormal mitotic exit after a short delay. Quantitative live-cell imaging approaches combined with mathematical modeling indicate that weak SAC activation upon cohesion loss is caused by weak signal generation. This is further attenuated by several feedback loops in the mitotic signaling network. We propose that multiple feedback loops involving cyclin-dependent kinase 1 (Cdk1) gradually impair error-correction efficiency and accelerate mitotic exit upon premature loss of cohesion. Our findings explain how cohesion defects may escape SAC surveillance.
-
Wednesday, Jul 1, 2015
BMC Biology
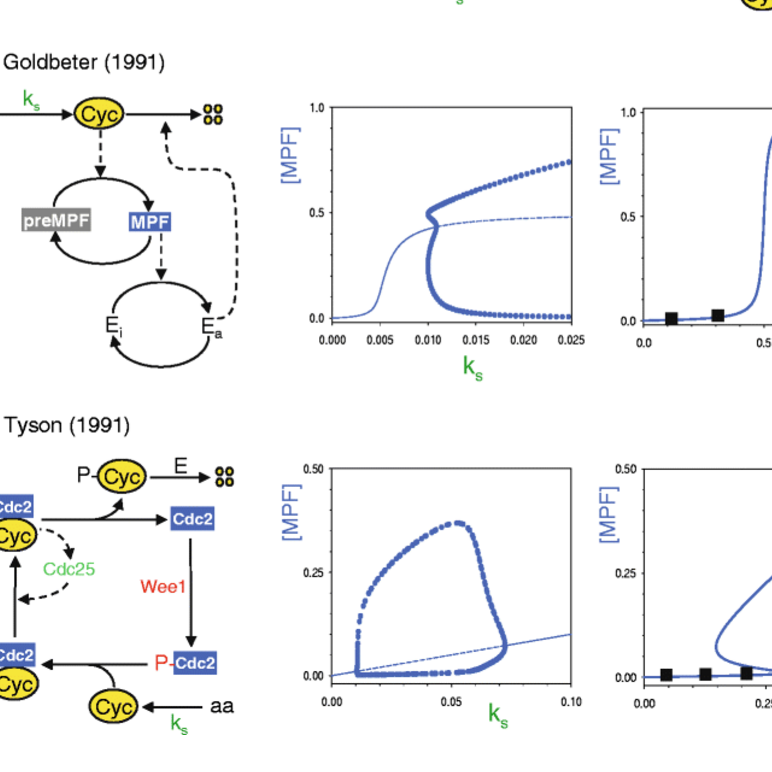
Models in biology: lessons from modeling regulation of the eukaryotic cell cycle
John Tyson, Bela Novak
-
AbstractIn this essay we illustrate some general principles of mathematical modeling in biology by our experiences in studying the molecular regulatory network underlying eukaryotic cell division. We discuss how and why the models moved from simple, parsimonious cartoons to more complex, detailed mechanisms with many kinetic parameters. We describe how the mature models made surprising and informative predictions about the control system that were later confirmed experimentally. Along the way, we comment on the ‘parameter estimation problem’ and conclude with an appeal for a greater role for mathematical models in molecular cell biology.
-
Thursday, Feb 12, 2015
FEBS Letters
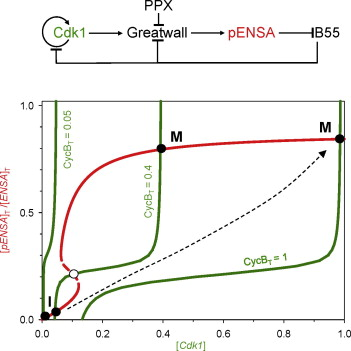
Model scenarios for switch-like mitotic transitions
Vinod PK, Bela Novak
-
AbstractTo facilitate rapid accumulation of Cdk1-phosphorylated substrate proteins, the Cdk1 counter-acting phosphatase, PP2A-B55 is inhibited during M phase by stoichiometric inhibitors (ENSA and Arpp19). These inhibitors are activated when phosphorylated by Cdk1-activated Greatwall-kinase. Recent experiments show that ENSA is dephosphorylated and inactivated by the PP2A-B55 itself, and acts as an unfair substrate inhibiting PP2A-B55 activity towards other Cdk1 substrates. Mathematical modelling shows that this mutual antagonism between the phosphatase and its inhibitor is insufficient to explain the switch-like characteristics of mitotic entry and exit. We show that the feedback regulation of Greatwall activating kinase and/or inactivating phosphatase can explain the abruptness of these cell cycle transitions.