Stefan Heldt
Postdoc

Stefan Heldt studied Biosystems Engineering at the Otto von Guericke University in Magdeburg, Germany. He received his degree in 2009 after completing a diploma thesis on stochastic simulations of mRNA translation in the group of Prof. Celso Grebogi at the University of Aberdeen, Scotland. He then joined the Max Planck Institute for Dynamics of Complex Technical Systems for a Ph.D. developing mechanistic, multi-scale models of influenza virus infection. This work awarded him the 2016 MTZ Award for Medical Systems Biology. Since 2015, he is a postdoctoral research fellow in Bela Novak’s lab, where he uses mathematical modelling to understand the dynamic characteristics of the mammalian cell cycle and its regulation. In this project, he is working closely with Alexis Barr from Chris Bakal’s lab.
Publications contributed to in the context of this project
-
Thursday, Jan 9, 2020
Current Biology
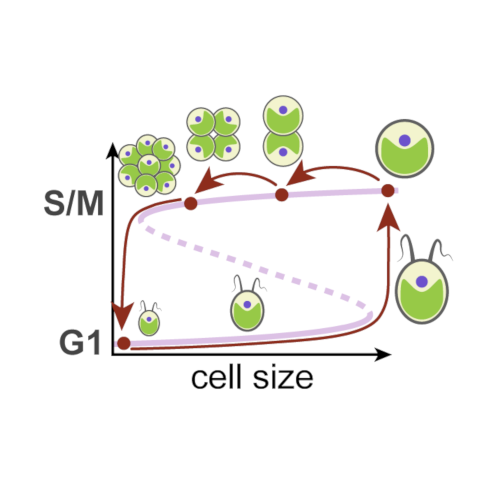
A single light-responsive sizer can control multiple-fission cycles in Chlamydomonas
Stefan Heldt, John J. Tyson, Frederick R. Cross, Bela Novak
-
AbstractMost eukaryotic cells execute binary division after each mass doubling in order to maintain size homeostasis by coordinating cell growth and division. By contrast, the photosynthetic green alga Chlamydomonas can grow more than 8-fold during daytime and then, at night, undergo rapid cycles of DNA replication, mitosis, and cell division, producing up to 16 daughter cells. Here, we propose a mechanistic model for multiple-fission cycles and cell-size control in Chlamydomonas. The model comprises a light-sensitive and size-dependent biochemical toggle switch that acts as a sizer, guarding transitions into and exit from a phase of cell-division cycle oscillations. This simple “sizer-oscillator” arrangement reproduces the experimentally observed features of multiple-fission cycles and the response of Chlamydomonas cells to different light-dark regimes. Our model also makes specific predictions about the size dependence of the time of onset of cell division after cells are transferred from light to dark conditions, and we confirm these predictions by single-cell experiments. Collectively, our results provide a new perspective on the concept of a “commitment point” during the growth of Chlamydomonas cells and hint at intriguing similarities of cell-size control in different eukaryotic lineages.
-
Wednesday, Oct 24, 2018
PLOS Computational Biology
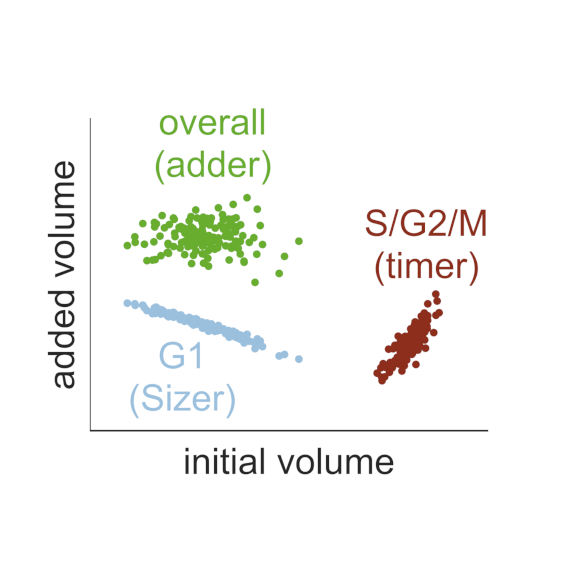
Dilution and titration of cell-cycle regulators may control cell size in budding yeast
Stefan Heldt, Reece Lunstone, John J. Tyson, Bela Novak
-
AbstractThe size of a cell sets the scale for all biochemical processes within it, thereby affecting cellular fitness and survival. Hence, cell size needs to be kept within certain limits and relatively constant over multiple generations. However, how cells measure their size and use this information to regulate growth and division remains controversial. Here, we present two mechanistic mathematical models of the budding yeast (S. cerevisiae) cell cycle to investigate competing hypotheses on size control: inhibitor dilution and titration of nuclear sites. Our results suggest that an inhibitor-dilution mechanism, in which cell growth dilutes the transcriptional inhibitor Whi5 against the constant activator Cln3, can facilitate size homeostasis. This is achieved by utilising a positive feedback loop to establish a fixed size threshold for the START transition, which efficiently couples cell growth to cell cycle progression. Yet, we show that inhibitor dilution cannot reproduce the size of mutants that alter the cell’s overall ploidy and WHI5 gene copy number. By contrast, size control through titration of Cln3 against a constant number of genomic binding sites for the transcription factor SBF recapitulates both size homeostasis and the size of these mutant strains. Moreover, this model produces an imperfect ‘sizer’ behaviour in G1 and a ‘timer’ in S/G2/M, which combine to yield an ‘adder’ over the whole cell cycle; an observation recently made in experiments. Hence, our model connects these phenomenological data with the molecular details of the cell cycle, providing a systems-level perspective of budding yeast size control.
-
Thursday, Feb 22, 2018
Current Opinion in Systems Biology
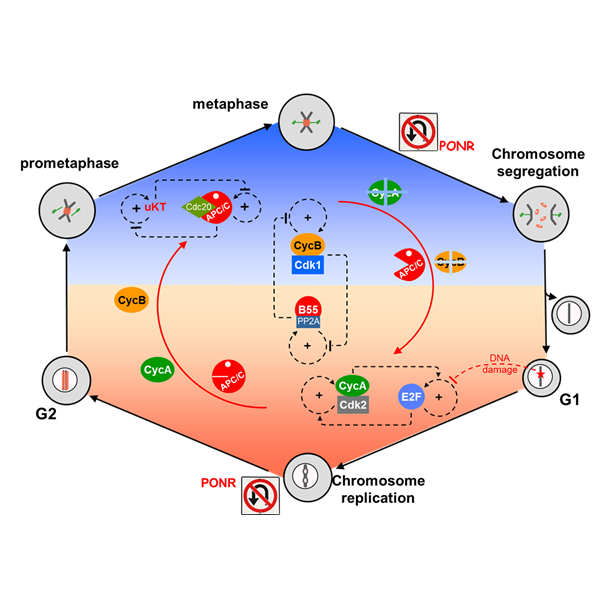
Genome Stability during Cell Proliferation: A Systems Analysis of the Molecular Mechanisms Controlling Progression through the Eukaryotic Cell Cycle
Bela Novak, Stefan Heldt, John J. Tyson
-
AbstractWell-nourished cells in a favorable environment (well supplied with growth factors, cytokines, and/or hormones and free from stresses, ionizing radiation, etc.) will grow, replicate their genome, and divide into two daughter cells, fully prepared to repeat the process. This cycle of DNA replication and division underlies all aspects of biological growth, reproduction, repair and development. As such, it is essential that the cell’s genome be guarded against damage during the replication/division process, lest the error(s) be irrevocably passed down to all future generations of progeny. Hence, cell cycle progression is closely guarded against major sources of errors, in particular DNA damage and misalignment of replicated chromosomes on the mitotic spindle. In this review article we examine closely the molecular mechanisms that maintain genomic integrity during the cell division cycle, and we find an unexpected and intriguing arrangement of concatenated and nested bistable toggle switches. The topology of the network seems to play crucial roles in maintaining the stability of the genome during cell proliferation.
-
Wednesday, Feb 21, 2018
Proceedings of the National Academy of Sciences
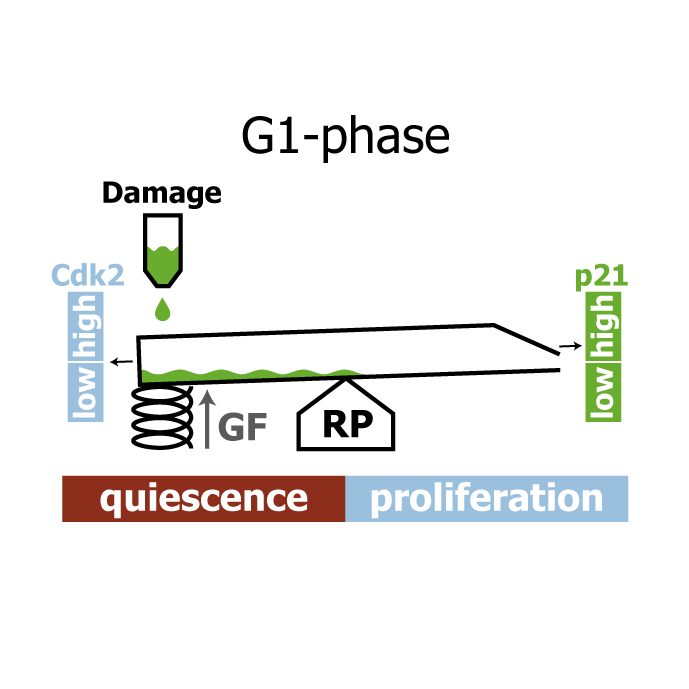
A comprehensive model for the proliferation-quiescence decision in response to endogenous DNA damage in human cells
Stefan Heldt, Alexis Barr, Sam Cooper, Chris Bakal, Bela Novak
-
AbstractHuman cells that suffer mild DNA damage can enter a reversible state of growth arrest known as quiescence. This decision to temporarily exit the cell cycle is essential to prevent the propagation of mutations and most cancer cells harbour defects in the underlying control system. Here we present a mechanistic mathematical model to study the proliferation-quiescence decision in non-transformed human cells. We show that two bistable switches, the restriction point (RP) and the G1/S transition, mediate this decision by integrating DNA damage and mitogen signals. In particular, our data suggests that the cyclin-dependent kinase inhibitor p21 (Cip1/Waf1), which is expressed in response to DNA damage, promotes quiescence by blocking positive feedback loops that facilitate G1 progression downstream of serum stimulation. Intriguingly, cells exploit bistability in the RP to convert graded p21 and mitogen signals into an all-or-nothing cell cycle response. The same mechanism creates a window of opportunity where G1 cells that have passed the RP can revert to quiescence if exposed to DNA damage. We present experimental evidence that cells gradually lose this ability to revert to quiescence as they progress through G1 and that the onset of rapid p21 degradation at the G1/S transition prevents this response altogether, insulating S-phase from mild, endogenous DNA damage. Thus, two bistable switches conspire in the early cell cycle to provide both sensitivity and robustness to external stimuli.
-
Monday, Mar 20, 2017
Nature Communications
DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression
Alexis Barr, Samuel Cooper, Stefan Heldt, Francesca Butera, Henriette Stoy, Jörg Mansfeld, Bela Novak, Chris Bakal
-
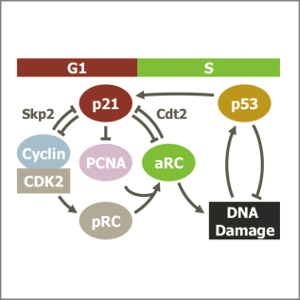
AbstractFollowing DNA damage caused by exogenous sources, such as ionising radiation, the tumour suppressor p53 mediates cell cycle arrest via expression of the CDK inhibitor, p21. However, the role of p21 in maintaining genomic stability in the absence of exogenous DNA damaging agents is unclear. Here, using live single cell measurements of p21 protein in proliferating cultures, we show that naturally-occurring DNA damage incurred over S-phase causes p53-dependent accumulation of p21 during mother G2- and daughter G1-phases. High p21 levels mediate G1 arrest via CDK inhibition, yet lower levels have no impact on G1 progression, and the ubiquitin ligases CRL4Cdt2 and SCFSkp2 couple to degrade p21 prior to the G1/S transition. Mathematical modelling reveals that a bistable switch, created by CRL4Cdt2, promotes irreversible S-phase entry by keeping p21 levels low, preventing premature S-phase exit upon DNA damage. Thus, we characterise how p21 regulates the proliferation-quiescence decision to maintain genomic stability.
-
Wednesday, Jan 27, 2016
Cell Systems
A Dynamical Framework for the All-or-None G1/S Transition
Alexis Barr, Stefan Heldt, Tongli Zhang, Chris Bakal, Bela Novak
-
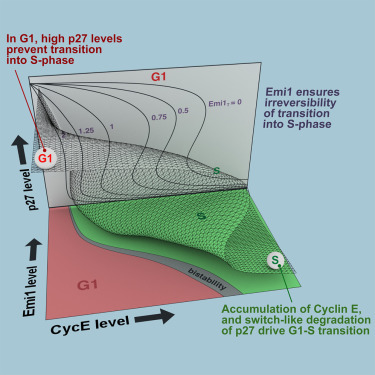
AbstractThe transition from G1 into DNA replication (S phase) is an emergent behavior resulting from dynamic and complex interactions between cyclin-dependent kinases (Cdks), Cdk inhibitors (CKIs), and the anaphase-promoting complex/cyclosome (APC/C). Understanding the cellular decision to commit to S phase requires a quantitative description of these interactions. We apply quantitative imaging of single human cells to track the expression of G1/S regulators and use these data to parametrize a stochastic mathematical model of the G1/S transition. We show that a rapid, proteolytic, double-negative feedback loop between Cdk2:Cyclin and the Cdk inhibitor p27Kip1 drives a switch-like entry into S phase. Furthermore, our model predicts that increasing Emi1 levels throughout S phase are critical in maintaining irreversibility of the G1/S transition, which we validate using Emi1 knockdown and live imaging of G1/S reporters. This work provides insight into the general design principles of the signaling networks governing the temporally abrupt transitions between cell-cycle phases.