Chris Bakal
Principal Investigator

Chris Bakal’s contributions to biology have been to use imaging and computational methods to describe the signalling networks regulating cell shape. Specifically, the Bakal laboratory has developed the use of Quantitative Morphological Signatures (QMSs) to describe the contribution of individual genes to the regulation of single-cell shape (Bakal et al., Science 2007), automated online phenotype discovery algorithms that search through databases containing millions of images to identify novel cell shapes (Yin et al., BMC Bioinformatics 2008), algorithms to determine hits from RNAi screens based on the known physical structure of signalling networks (Kraplow et al., Nature Methods 2009), combinatorial RNAi methods to determine epistatic relationships in metazoan systems (Arias-Garcia et al., Molecular Biosystems 2012), methods to describe networks through integration of phosphoproteomic and functional genomic data (Bakal et al., Science 2008), and algorithms to model hierarchical signalling relationships from QMSs derived from combinatorial RNAi (Nir et al., Genome Research 2010). Recently the Bakal laboratory devised methods to quantify the morphological heterogeneity of populations in the context of a high-throughput genetic screen, and identified a signalling network that regulates the switch-like changes in cell shape made by melanoma cells during the metastatic process (Yin et al., Nature Cell Biology 2013). The Bakal laboratory will focus on G1/S transition and will be responsible for the live cell imaging facilities as well.
Publications contributed to in the context of this project
-
Thursday, Apr 30, 2020
EMBO Journal
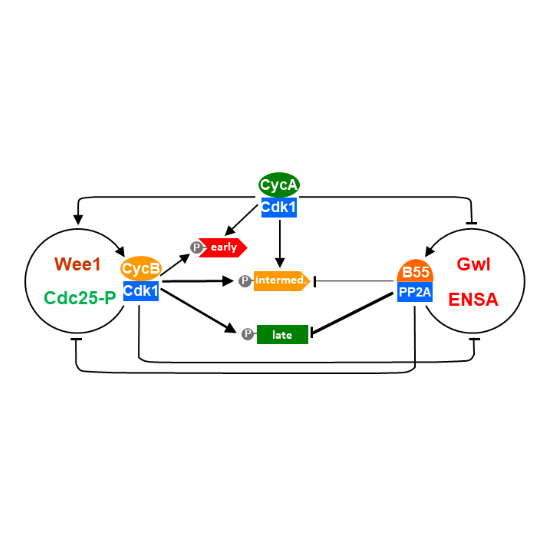
Cyclin A Triggers Mitosis Either via the Greatwall Kinase Pathway or Cyclin B
Nadia Hegarat, Adrijana Crncec, Maria F Suarez Peredo Rodriguez, Fabio Echegaray Iturra, Yan Gu, Oliver Busby, Paul F Lang, Alexis R Barr, Chris Bakal, Masato T Kanemaki, Angus I Lamond, Bela Novak, Tony Ly, Helfrid Hochegger
-
AbstractTwo mitotic cyclin types, cyclin A and B, exist in higher eukaryotes, but their specialised functions in mitosis are incompletely understood. Using degron tags for rapid inducible protein removal, we analyse how acute depletion of these proteins affects mitosis. Loss of cyclin A in G2-phase prevents mitotic entry. Cells lacking cyclin B can enter mitosis and phosphorylate most mitotic proteins, because of parallel PP2A:B55 phosphatase inactivation by Greatwall kinase. The final barrier to mitotic establishment corresponds to nuclear envelope breakdown, which requires a decisive shift in the balance of cyclin-dependent kinase Cdk1 and PP2A:B55 activity. Beyond this point, cyclin B/Cdk1 is essential for phosphorylation of a distinct subset of mitotic Cdk1 substrates that are essential to complete cell division. Our results identify how cyclin A, cyclin B and Greatwall kinase coordinate mitotic progression by increasing levels of Cdk1-dependent substrate phosphorylation.
-
Wednesday, Feb 21, 2018
Proceedings of the National Academy of Sciences
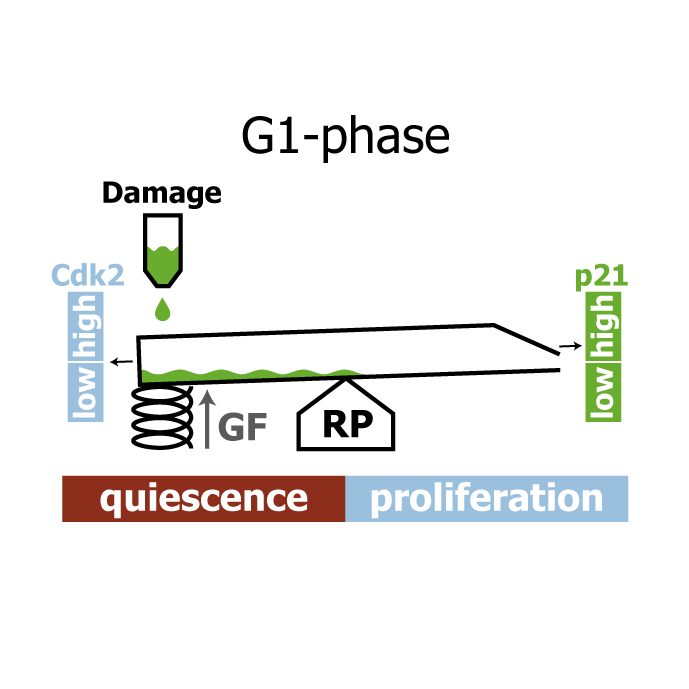
A comprehensive model for the proliferation-quiescence decision in response to endogenous DNA damage in human cells
Stefan Heldt, Alexis Barr, Sam Cooper, Chris Bakal, Bela Novak
-
AbstractHuman cells that suffer mild DNA damage can enter a reversible state of growth arrest known as quiescence. This decision to temporarily exit the cell cycle is essential to prevent the propagation of mutations and most cancer cells harbour defects in the underlying control system. Here we present a mechanistic mathematical model to study the proliferation-quiescence decision in non-transformed human cells. We show that two bistable switches, the restriction point (RP) and the G1/S transition, mediate this decision by integrating DNA damage and mitogen signals. In particular, our data suggests that the cyclin-dependent kinase inhibitor p21 (Cip1/Waf1), which is expressed in response to DNA damage, promotes quiescence by blocking positive feedback loops that facilitate G1 progression downstream of serum stimulation. Intriguingly, cells exploit bistability in the RP to convert graded p21 and mitogen signals into an all-or-nothing cell cycle response. The same mechanism creates a window of opportunity where G1 cells that have passed the RP can revert to quiescence if exposed to DNA damage. We present experimental evidence that cells gradually lose this ability to revert to quiescence as they progress through G1 and that the onset of rapid p21 degradation at the G1/S transition prevents this response altogether, insulating S-phase from mild, endogenous DNA damage. Thus, two bistable switches conspire in the early cell cycle to provide both sensitivity and robustness to external stimuli.
-

Tuesday, Jun 20, 2017
Bioinformatics
NucliTrack: An integrated nuclei tracking application
Sam Cooper, Alexis Barr, Robert Glen, Chris Bakal
-
AbstractLive imaging studies give unparalleled insight into dynamic single cell behaviours and fate decisions. However, the challenge of reliably tracking single cells over long periods of time limits both the throughput and ease with which such studies can be performed. Here, we present NucliTrack, a cross platform solution for automatically segmenting, tracking and extracting features from fluorescently-labelled nuclei. NucliTrack performs similarly to other state-of-the-art cell tracking algorithms, but NucliTrack’s interactive, graphical interface makes it significantly more user friendly.
-
Monday, Mar 20, 2017
Nature Communications
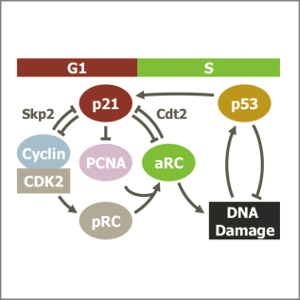
DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression
Alexis Barr, Samuel Cooper, Stefan Heldt, Francesca Butera, Henriette Stoy, Jörg Mansfeld, Bela Novak, Chris Bakal
-
AbstractFollowing DNA damage caused by exogenous sources, such as ionising radiation, the tumour suppressor p53 mediates cell cycle arrest via expression of the CDK inhibitor, p21. However, the role of p21 in maintaining genomic stability in the absence of exogenous DNA damaging agents is unclear. Here, using live single cell measurements of p21 protein in proliferating cultures, we show that naturally-occurring DNA damage incurred over S-phase causes p53-dependent accumulation of p21 during mother G2- and daughter G1-phases. High p21 levels mediate G1 arrest via CDK inhibition, yet lower levels have no impact on G1 progression, and the ubiquitin ligases CRL4Cdt2 and SCFSkp2 couple to degrade p21 prior to the G1/S transition. Mathematical modelling reveals that a bistable switch, created by CRL4Cdt2, promotes irreversible S-phase entry by keeping p21 levels low, preventing premature S-phase exit upon DNA damage. Thus, we characterise how p21 regulates the proliferation-quiescence decision to maintain genomic stability.
-
Wednesday, Jan 27, 2016
Cell Systems
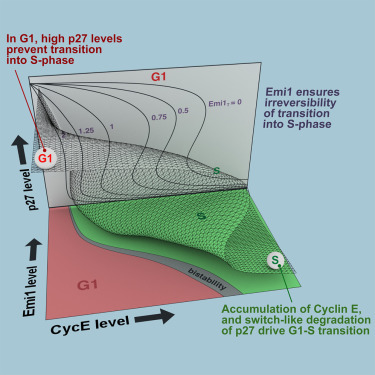
A Dynamical Framework for the All-or-None G1/S Transition
Alexis Barr, Stefan Heldt, Tongli Zhang, Chris Bakal, Bela Novak
-
AbstractThe transition from G1 into DNA replication (S phase) is an emergent behavior resulting from dynamic and complex interactions between cyclin-dependent kinases (Cdks), Cdk inhibitors (CKIs), and the anaphase-promoting complex/cyclosome (APC/C). Understanding the cellular decision to commit to S phase requires a quantitative description of these interactions. We apply quantitative imaging of single human cells to track the expression of G1/S regulators and use these data to parametrize a stochastic mathematical model of the G1/S transition. We show that a rapid, proteolytic, double-negative feedback loop between Cdk2:Cyclin and the Cdk inhibitor p27Kip1 drives a switch-like entry into S phase. Furthermore, our model predicts that increasing Emi1 levels throughout S phase are critical in maintaining irreversibility of the G1/S transition, which we validate using Emi1 knockdown and live imaging of G1/S reporters. This work provides insight into the general design principles of the signaling networks governing the temporally abrupt transitions between cell-cycle phases.